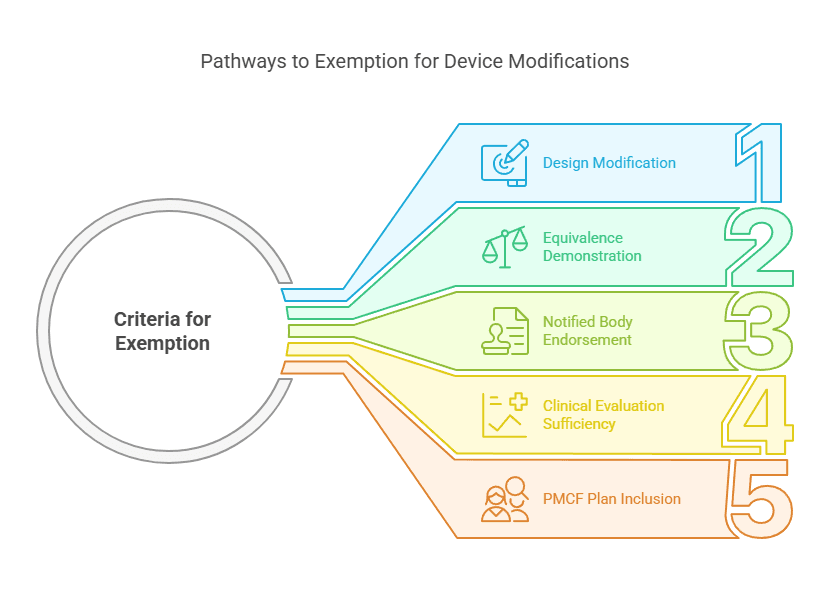
Cas n°1 : Modification d’un Dispositif déjà Commercialisé par le Même Fabricant
Lorsque le dispositif évalué (DUE) est une version modifiée d’un dispositif déjà commercialisé par le même fabricant, la réalisation d’une nouvelle investigation clinique peut ne pas être exigée.
Critères de dérogation :
Le DUE correspond à une modification de conception d’un dispositif existant du même fabricant.
L’équivalence avec le dispositif commercialisé est démontrée conformément à l’annexe XIV, section 3.
L’organisme notifié valide l’argumentaire d’équivalence.
L’évaluation clinique du dispositif déjà commercialisé est suffisante pour démontrer la conformité aux exigences générales en matière de sécurité et de performance (GSPR).
Le plan de suivi clinique après commercialisation (SCAC/PMCF) inclut des études post-commercialisation visant à vérifier la sécurité et la performance du dispositif.
Référence :
Article 61(4) du MDR, premier à troisième tirets
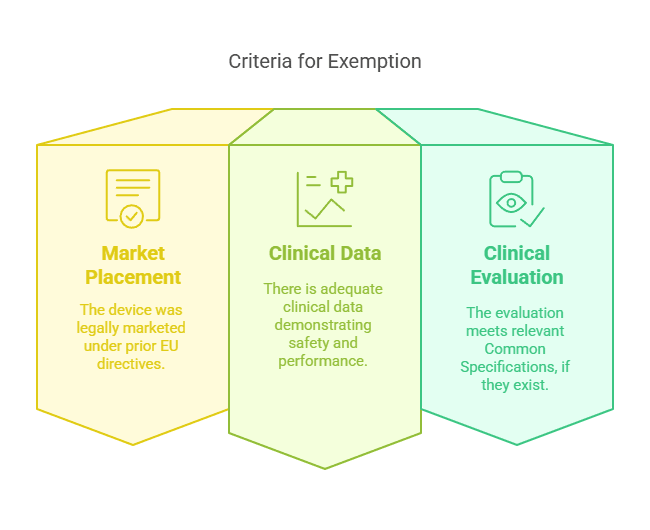
Cas n°2 : Dispositifs « Legacy » Mis sur le Marché selon les Anciennes Directives
Les dispositifs ayant été légalement mis sur le marché conformément à la directive 90/385/CEE ou à la directive 93/42/CEE avant l’entrée en application du MDR peuvent être exemptés d’investigation clinique, à condition que des données cliniques suffisantes soient disponibles.
Critères de dérogation :
Le DUE a été mis sur le marché légalement sous l’empire des anciennes directives européennes.
Des données cliniques suffisantes étayent la sécurité et la performance du dispositif.
L’évaluation clinique est conforme aux Spécifications Communes (CS) applicables, si elles existent.
Référence :
Article 61(6)(a) du MDR
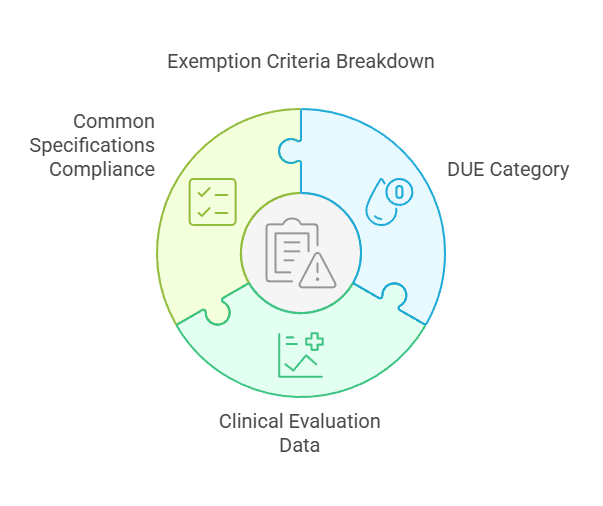
Cas n°3 : Technologie Bien Établie (WET – Well-Established Technology)
Les dispositifs relevant d’une technologie bien établie (Well-Established Technology), tels que les sutures, agrafes, vis ou obturations dentaires, peuvent être exemptés d’une investigation clinique si des données cliniques existantes sont suffisantes.
Critères de dérogation :
Le DUE appartient à la catégorie des technologies bien établies (WET).
L’évaluation clinique est étayée par des données cliniques suffisantes.
La conformité aux Spécifications Communes applicables est assurée, si elles existent.
Référence :
Article 61(6)(b) du MDR
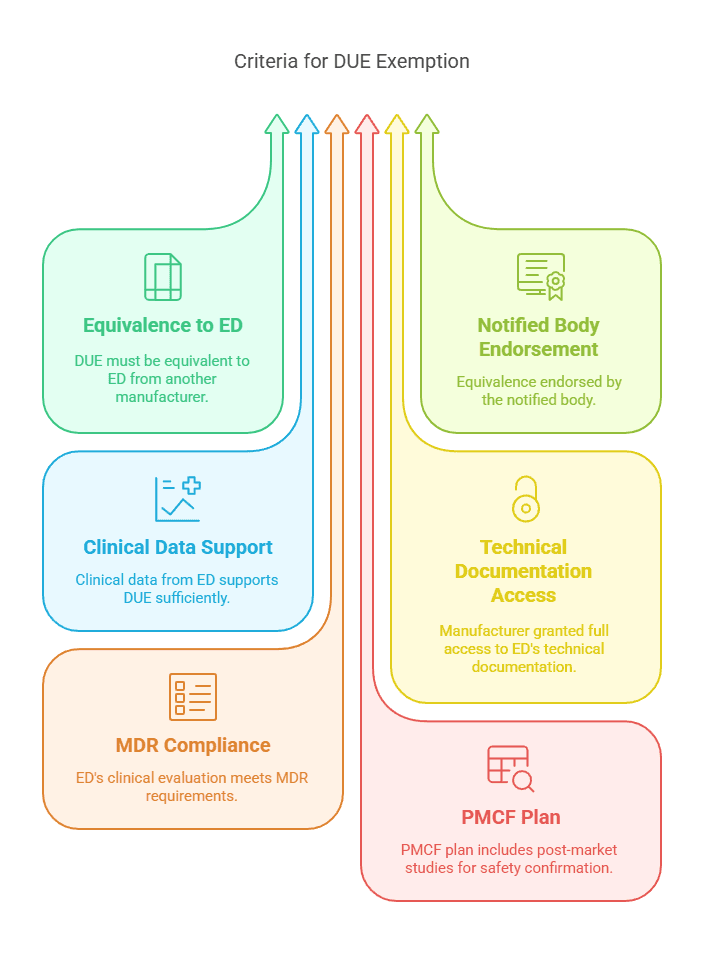
Cas n°4 : Dispositif Équivalent d’un Autre Fabricant
Un fabricant peut éviter de réaliser une investigation clinique pour un dispositif médical en démontrant l’équivalence avec un dispositif équivalent (ED) d’un autre fabricant, à condition de disposer d’un accès complet à la documentation technique de l’ED via un accord contractuel.
Critères de dérogation :
Le DUE est équivalent à un dispositif d’un autre fabricant, conformément à l’annexe XIV, section 3.
L’équivalence est validée par l’organisme notifié.
Les données cliniques issues de l’ED sont suffisantes pour étayer la sécurité et la performance du DUE.
Un contrat accorde explicitement au fabricant un accès complet à la documentation technique de l’ED.
L’évaluation clinique de l’ED est conforme aux exigences du MDR.
Le plan de suivi clinique après commercialisation (SCAC/PMCF) comprend des études post-commercialisation visant à confirmer la sécurité et la performance.
Référence :
Article 61(5) du MDR