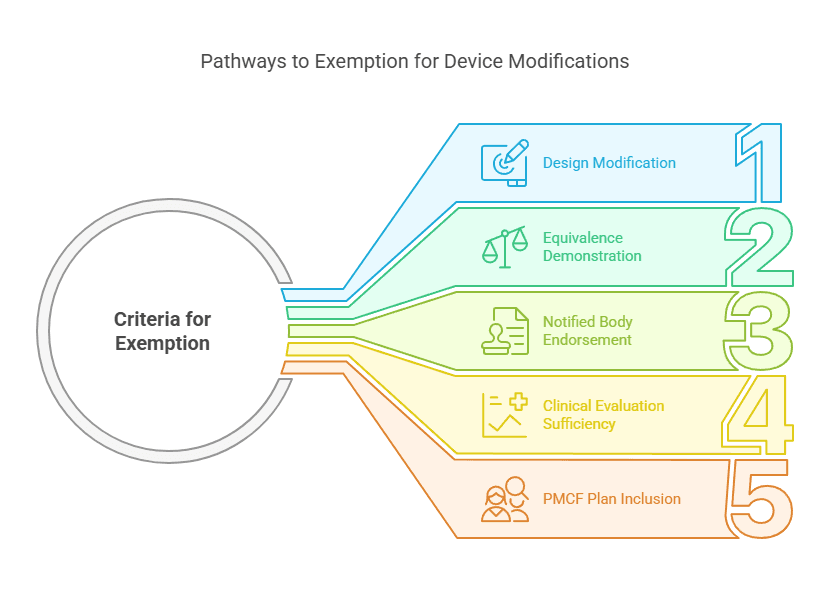
Case 1: Modification of a Marketed Device by the Same Manufacturer
If the Device Under Evaluation (DUE) is a modified version of an already marketed device from the same manufacturer, a new clinical investigation may not be required.
Criteria for Exemption:
The DUE is a design modification of an existing device from the same manufacturer.
Equivalence with the marketed device is demonstrated per Annex XIV Section 3.
The notified body endorses the equivalence claim.
The clinical evaluation of the marketed device is sufficient to demonstrate conformity with General Safety and Performance Requirements (GSPRs).
A Post-Market Clinical Follow-Up (PMCF) plan includes post-market studies to verify safety and performance.
Reference:
MDR Article 61(4), Indents 1-3
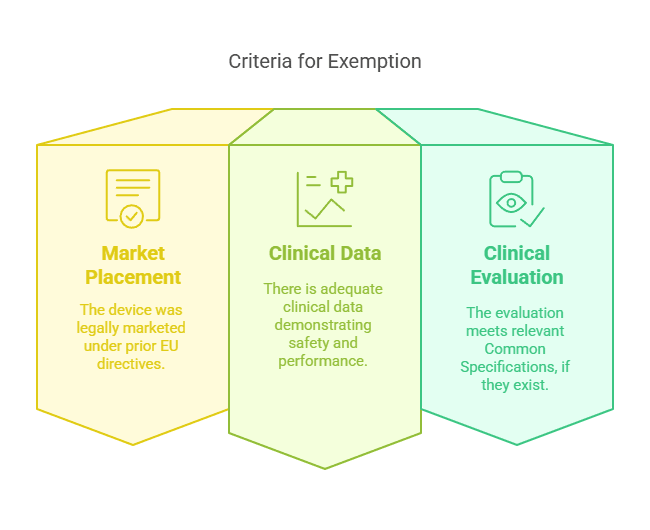
Case 2: Legacy Devices Placed on the Market Under Previous Directives
Devices that were legally placed on the market under Directive 90/385/EEC or Directive 93/42/EEC before the MDR came into effect may be exempt from clinical investigations if sufficient clinical data is available.
Criteria for Exemption:
The DUE was lawfully placed on the market under previous EU directives.
Sufficient clinical data supports the safety and performance of the device.
The clinical evaluation aligns with applicable Common Specifications (CS), if available.
Reference:
MDR Article 61(6)(a)
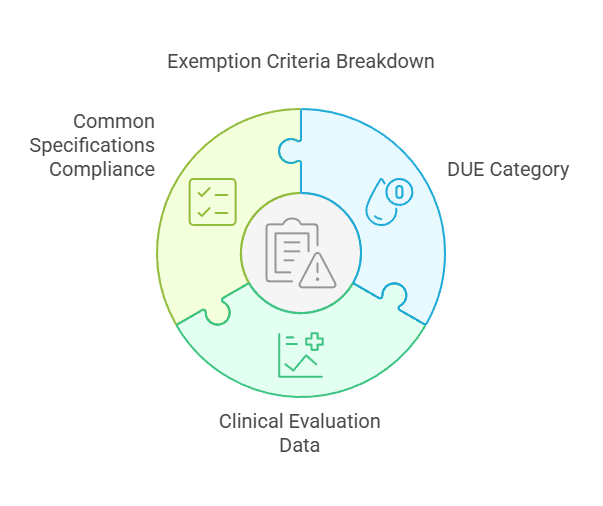
Well-Established Technology (WET) devices, such as sutures, staples, screws, and dental fillings, may avoid clinical investigation for medical devices if existing clinical data is sufficient.
Criteria for Exemption:
The DUE falls within the category of WET.
The clinical evaluation is supported by sufficient clinical data.
Compliance with relevant Common Specifications is ensured, if applicable.
Reference:
MDR Article 61(6)(b)
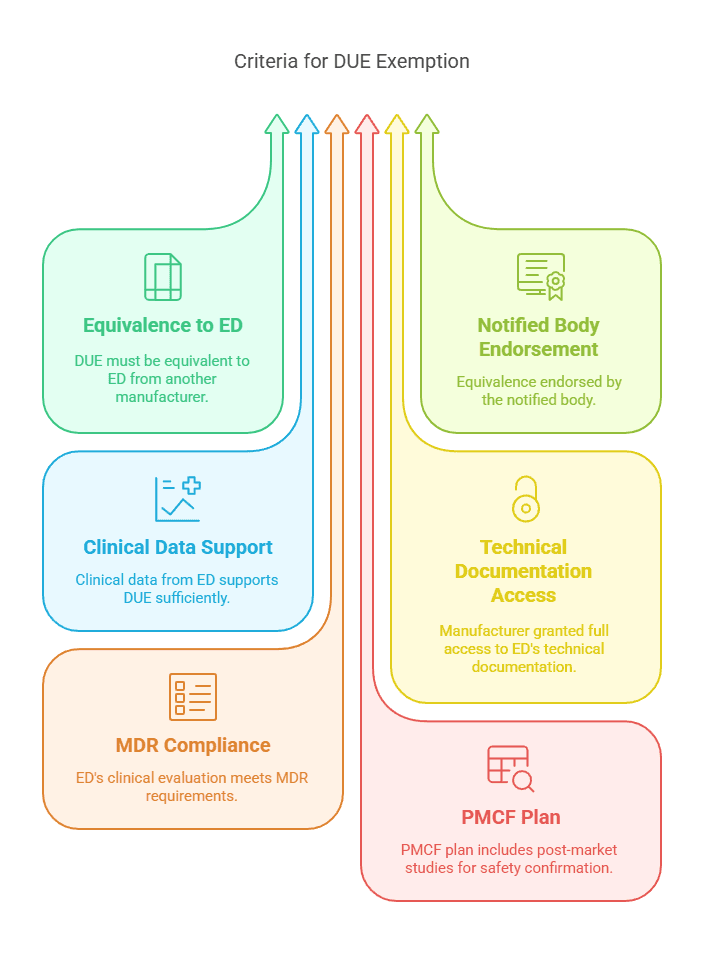
Case 4: Equivalent Device from Another Manufacturer
A manufacturer may avoid clinical investigation for a medical device by demonstrating equivalence to another manufacturer’s device, provided that full access to the technical documentation of the equivalent device (ED) is ensured through a contractual agreement.
Criteria for Exemption:
The DUE is equivalent to an ED from another manufacturer per Annex XIV Section 3.
Equivalence is endorsed by the notified body.
Clinical data from the ED sufficiently supports the DUE.
A contract explicitly grants the manufacturer full access to the ED’s technical documentation.
The ED’s clinical evaluation meets MDR requirements.
A PMCF plan includes post-market studies to confirm safety and performance.
Reference:
MDR Article 61(5)